
Genetic diversity of UK honey bees
The original population of honey bees found in the UK was the subspecies Apis mellifera mellifera (A.m.m.), the dark honey bee.
Imports of Italian and Carniolan honey bees began in the nineteenth century and increased during the twentieth century, resulting in widespread interbreeding with the native population.
Brother Adam claimed that his C-lineage honey bees did not succumb to Isle of Wight disease, and began producing his first Buckfast strain in 1919 by crossing A.m.m. with Italian honey bees at Buckfast Abbey in Devon. Using other subspecies he went on to breed these onto his now famous Buckfast lines. If we assume that Isle of Wight disease was a combination of chronic bee paralysis virus, poor weather and lack of forage (Neumann and Carreck, 2010), little is known currently about resistant strains. It is likely that within A.m. ligustica and A.m. carnica there are strains both susceptible and resistant to chronic bee paralysis virus.
Structure and function of DNA
DNA (deoxyribonucleic acid), found in the nucleus of every cell, is composed of two molecular chains which coil around each other to form a double helix.
It carries genetic instructions used by an organism to control its growth, development and bodily functions. Each chain, or strand, is comprised of four different ‘nucleotides’ often referred to by the first letter of their names: A (adenine), T (thymine), G (guanine) or C (cytosine).
The specific order or sequence of these four nucleotides forms a genetic code. Specific lengths of this code form genes which contain the information used by the cell to make the many different proteins an organism needs to survive and prosper.
Sequencing DNA can be used to learn more about an organism, the relationship of individuals of a species to each other and to other species, what proteins they make and how changes in the code may make them better or worse at adapting to new conditions, whether environmental challenges or attack by pathogens.
Quicker and cheaper
There has been a revolution in DNA sequencing technology in the past two decades. So-called Next Generation Sequencing allows biologists to sequence DNA much more quickly and cheaply than ever before.
For example, the human genome project, which was completed in 2001, took around ten years to complete and cost three billion dollars; today it costs less than $1000 and takes only three days to sequence a human genome.
By comparison, the genome of the honey bee is 10% the size of a human’s and it is therefore now becoming cost-effective to analyse the genetic make-up of bee populations using this approach.
Whole genome sequencing
At the Roslin Institute in Edinburgh, we have performed a pilot project to begin to study the genetic diversity of honey bees in the UK using whole genome sequencing. Pools of 16 worker bees were collected from 19 colonies in Scotland and England. From the Scottish population, we sampled 12 colonies, essentially at random, a colony of A.m.m. from the Isle of Colonsay, a Buckfast colony and a Carniolan colony. Three samples of A.m.m. were sampled from Norfolk, Warwickshire and Cambridgeshire together with a sample from an A.m.m. conservation project in Shropshire.
DNA was extracted from the samples and submitted to the Edinburgh Genomics facility for sequencing. Four of the samples were sequenced at two different depths (50X and 25X coverage, ie, each nucleotide in the genome is sequenced an average of 50 or 25 times). This was done to give an idea of the level or ‘depth’ of sequencing that would be optimal for analysing the UK population. The DNA sequences from each sample were lined up with the Apis mellifera reference genome and the genetic variation present in each colony was identified.
Identifying subspecies
Very common forms of genetic variation are called ‘small nucleotide variations’ (SNVs), where a single nucleotide in the DNA has been exchanged for a different one. Other commonly found variations between genomes are small insertions or deletions of nucleotide sequences. Using these genetic variations between colonies’ DNA sequences, we can identify the different subspecies of honey bee.
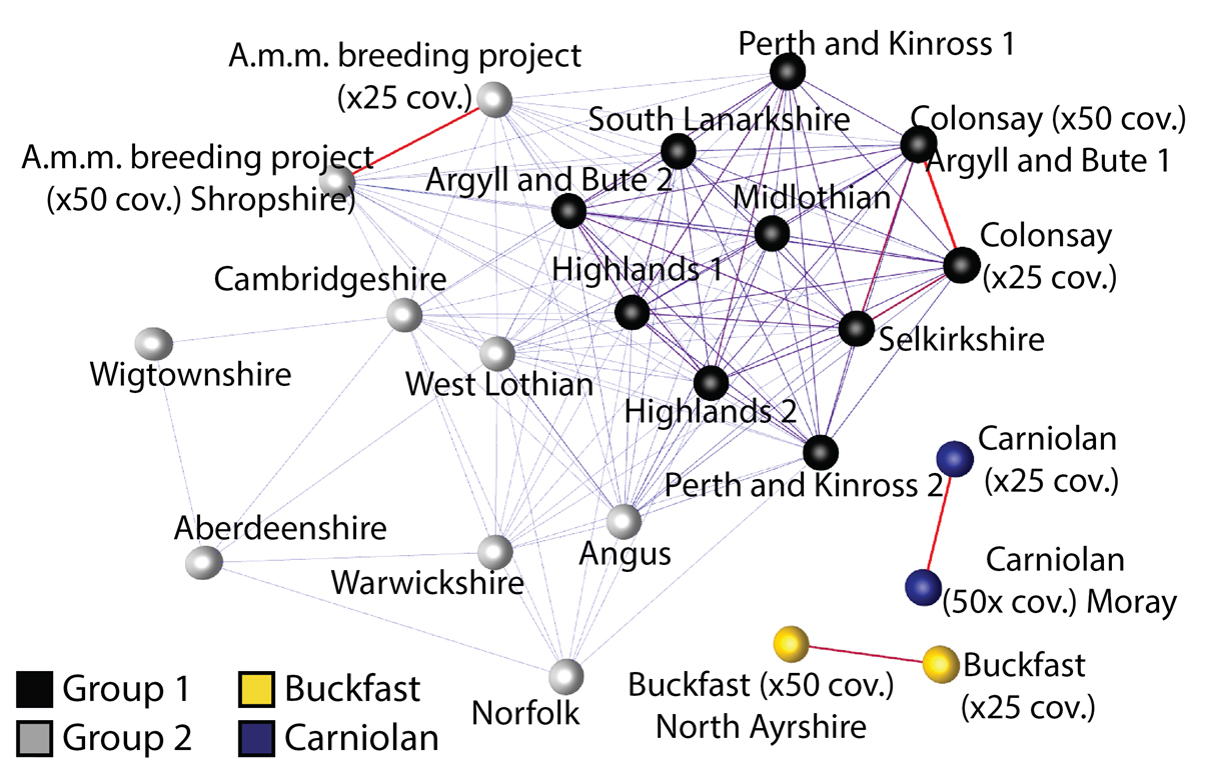
The figure shows a comparison of all the genetic variation across each colony (a similarity network). Each colony is represented by a sphere and the more similar one colony is to another, the thicker the line connecting them. The Carniolan sample and Buckfast sample are not connected to any other samples and their positioning on the network is arbitrary. If we progressively lowered the minimum similarity score used to construct the network, they would connect to each other and then to the Wigtownshire and Aberdeenshire samples.
Samples from the same colonies that were sequenced to different depths (25X and 50X coverage) are connected by very thick red lines, as would be expected. Many of the randomly sampled Scottish colonies are connected to the Colonsay sample (Group 1), bees that are known to be good examples of the native dark honey bee subspecies (A.m.m.). Interestingly, the examples of ‘A.m.m.’ sampled from England and the A.m.m. breeding project seemed to contain a higher proportion of Italian and Carniolan DNA than many of the randomly sampled Scottish samples. This suggests that A.m.m. might have a selective advantage in Scotland because of the climate or that A.m.m. populations are less affected by imported bees than those in England.
Although the sample size studied here is too small to draw any firm conclusions, this observation was supported in another study in which we sampled 96 colonies in Scotland and England using a genotyping platform developed by Dr Alice Pinto’s group in Portugal to estimate C-lineage introgression in the dark honey bee (Henriques, et al, 2018). This genotyping platform analyses 117 small nucleotide polymorphisms (SNPs) that have been selected to be most different between M-lineage (dark bee) and C-lineage (Italians and Carniolans).
Conclusions
In conclusion, whole genome sequencing is now a cost-effective means of studying honey bee genetics. It can help with conservation by identifying good examples of subspecies, thereby helping to maintain greater genetic diversity (the more genetic diversity within a species the better the chance it can adapt to the challenges of the modern environment).
Furthermore, analysis of the genetic differences in honey bees will help scientists to look for the regions of DNA involved in disease resistance or underlying different behaviours, such as spring build-up, swarming, temperament, and so on.
Finally, when you sequence a honey bee, you also sequence everything else living within the bee: its so-called microbiome, made up of ‘good’ bacteria such as those that help with the digestion of pollen, as well as any unwanted pests or pathogens (Regan, et al, 2018). We are beginning to use these rich data sets to help improve disease diagnostics and are hoping to identify beneficial microbial communities that help protect the bees against disease. In this way, whole genome sequencing can provide a comprehensive readout of a colony’s ancestry, as well as a health check on your bees.
Glossary
C-lineage: subspecies of Apis mellifera east and south of the Alps including those along the northern Mediterranean (A.m. carnica, A.m. cecropia, A.m. ligustica and A.m. macedonica).
Genetic diversity: the variation in the amount of genetic information within a population or a species.
Genome: the complete set of genetic material present in a cell or organism.
Introgression: movement of genes from one honey bee subspecies into the gene pool of another.
M-lineage: subspecies of Apis mellifera originating from western and northern Europe (A.m. mellifera and A.m. iberiensis). There has been a report of a recently discovered subspecies in China, A.m. sinisxinyuan (Chen, et al [2016]. Molecular Biology and Evolution, 33[5], 1337–1348).
Microbiome: the microorganisms found inside the bee.
Nucleotide: a nucleotide is one of the building blocks of DNA or RNA. A nucleotide consists of a base (adenine, thymine, guanine or cytosine) plus a molecule of sugar and one of phosphoric acid.
SNP: single nucleotide polymorphism is a substitution of a single nucleotide that occurs at a specific position in the genome, where each variation is present to some appreciable degree within a population (eg, >1%).
SNV: single nucleotide variation is a substitution of a single nucleotide that occurs at a specific position in the genome with no limitations placed on frequency by the researcher.
References
Neumann, P and Carreck, NL (2010). Honey bee colony losses.
Journal of Apicultural Research, 49(1), 1–6.
https://doi.org/10.3896/IBRA.1.49.1.01
Henriques, D, et al (2018). High sample throughput genotyping for estimating C-lineage introgression in the dark honeybee: an accurate and cost-effective SNP-based tool. Scientific Reports, 8(1), Article number 8552. www.nature.com/articles/s41598-018-26932-1
Regan, T*, Barnett, MW,* et al, Characterisation of the British honey bee metagenome. Nature Communications, 9(1), 4995. www.nature.com/articles/s41467-018-07426-0
* These authors contributed equally.
Written by Dr Mark W Barnett, Dr Tim Regan and Professor Tom C Freeman. This article appears in the June 2019 edition of Bee Craft magazine.